Zurück zur alphabetischen Auswahl
Humane spongiforme Enzephalopathie (HSE)
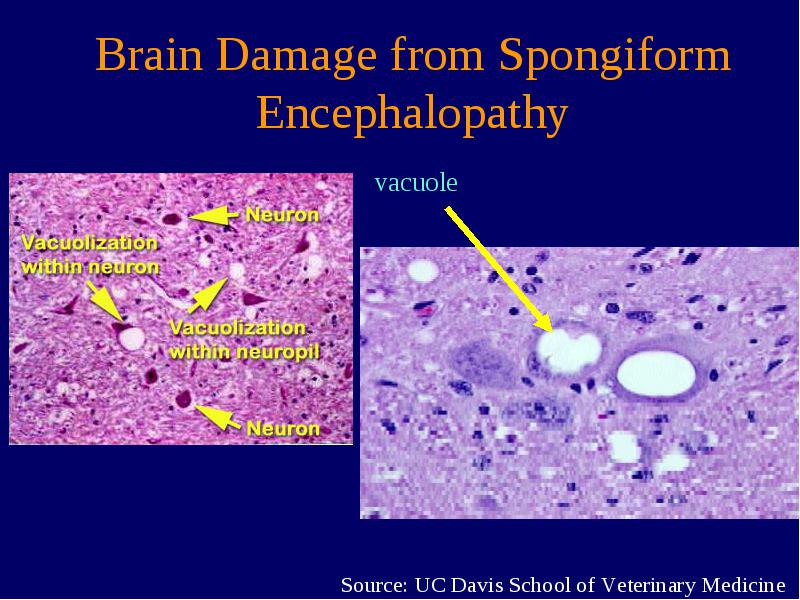
Creutzfeldt-Jakob-Krankheit (CJK) ist eine weitere Bezeichnung für die humane spongiforme Enzephalopathie. Die humane spongiforme Enzephalopathie ist eine degenerative, nicht entzündliche Erkrankung des zentralen Nervensystems, die zu schwammartigen (spongiformen) Gehirnerkrankungen (Enzephalopathien) führt. Die Erkrankung führt sehr schnell zu einer Demenz. Der Verlauf der Krankheit ist sehr unterschiedlich. Es kann der Tod schon nach wenigen Monaten eintreten, aber auch Verläufe über mehrere Jahre sind beschrieben worden. Es entstehen dabei fehlerhafte Proteine, die innerhalb des Gehirns zu Plaquebildung führen. Es kommt dadurch zum Untergang der Neurone.
Leitmerkmale: Gedächtnis-/Konzentrations-/Koordinationsstörungen, Schwindel, Krämpfe
| Definition | Bei der humanen spongiformen Enzephalopathie handelt es sich um einen immer weiter fortschreitenden Abbau von Zellen des zentralen Nervensystems |
| Weitere Bezeichnungen (Synonyme) |
|
| Formen |
|
| Erreger |
|
| Ausbreitung |
|
| Ansteckung |
|
| Kurzbeschreibung | Durch die Aufnahme der Prionen kommt es zu schwammigen Gehirnerkrankungen (graue Substanz), die durch massiven Zelluntergang gekennzeichnet sind. Dazu lagern sich die Prionen klumpenförmig zusammen und behindern so die Funktionen der Gehirnzellen Kennzeichen von Prionenerkrankungen:
|
| Inkubationszeit | 6 Monate-3 Jahre |
| Symptome |
|
| Diagnose | In der Inkubationszeit ist keine Diagnostik möglich: Anamnese: Verdachtsabklärung durch Symptomatik Apparative Diagnostik: Untersuchungen der Hirnflüssigkeit, EEG, Kernspin, evtl. Nachweis des veränderten Prion-Proteins (auch in Tonsillen, Appendix) |
| Differentialdiagnose |
|
| Komplikationen |
|
| Immunität/Prophylaxe |
|
| Therapie |
|
| Meldepflicht |
|
| Bilder |
{kind=link}
{kind=link}
ff